
Переломы при несовершенном остеогенезе

Несовершенный остеогенез – генетически обусловленная патология опорно-двигательного аппарата, характеризующаяся хрупкостью костной ткани и подверженностью ребенка частым переломам при минимальном воздействии или в отсутствие травмы. Кроме патологических переломов, при несовершенном остеогенезе у детей отмечаются деформации костей, аномалии зубов, атрофия мышц, гипермобильность суставов, прогрессирующая тугоухость. Диагноз несовершенного остеогенеза устанавливается с учетом анамнестических, клинических, рентгенологических данных, генетического тестирования. Лечение несовершенного остеогенеза включает профилактику переломов, бальнеотерапию, массаж, гимнастику, УФО, прием витамина D, препаратов кальция, фосфора, применение бисфосфонатов; при переломах – репозицию и гипсовую фиксацию отломков.
Общие сведения
Несовершенный остеогенез – наследственная патология, в основе которой лежит нарушение костеобразования (остеогенеза), приводящее к генерализованному остеопорозу и повышенной ломкости костей. Несовершенный остеогенез известен в литературе под различными названиями: врожденная хрупкость костей, внутриутробный рахит, периостальная дистрофия, болезнь Лобштейна (Фролика), врожденная остеомаляция и др. Из-за повышенной хрупкости костей и склонности к множественным переломам детей, страдающих несовершенным остеогенезом, часто называют «хрустальными детьми». Несовершенный остеогенез встречается с частотой 1 случай на 10 000-20 000 новорожденных. Несмотря на то, что как любое генетическое заболевание, несовершенный остеогенез неизлечим, сегодня существует возможность значительно облегчить и даже нормализовать жизнь «хрупких детей».
Несовершенный остеогенез
Причины несовершенного остеогенеза
Развитие несовершенного остеогенеза связано с врожденным нарушением обмена белка соединительной ткани коллагена 1-го типа, обусловленным мутациями генов, кодирующих коллагеновые цепи. В зависимости от формы заболевание может наследоваться по аутосомно-доминантному или аутосомно-рецессивному типу (менее 5%). Примерно в половине случаев патология возникает вследствие спонтанных мутаций. При несовершенном остеогенезе нарушается структура коллагена, входящего в состав костей и других соединительных тканей, либо синтезируется его недостаточное количество.
Нарушение синтеза коллагена остеобластами приводит к тому, что, несмотря на нормальный эпифизарный рост кости, нарушается периостальное и эндостальное окостенение. Костная ткань имеет пористое строение, состоит из костных островков и многочисленных пазух, заполненных рыхлой соединительной тканью; кортикальный слой истончен. Это обусловливает снижение механических свойств и патологическую ломкость костей при несовершенном остеогенезе.
Классификация несовершенного остеогенеза
Согласно классификации Д.О. Сайлленса, 1979 г., выделяют 4 генетических типа несовершенного остеогенеза:
I тип – имеет аутосомно-доминантное наследование, легкое или среднетяжелое течение. Характерны переломы умеренной тяжести, остеопороз, голубые склеры, ранняя тугоухость; несовершенный дентиногенез (подтип IA), без него – подтип IB.
II тип – предполагает аутосомно-рецессивное наследование, тяжелую перинатально-летальную форму. Оссификация черепа отсутствует, ребра имеют четкообразную форму, длинные трубчатые кости деформированы, емкость грудной клетки уменьшена. Множественные переломы костей возникают внутриутробно.
III тип – имеет аутосомно-рецессивное наследование. Протекает с тяжелой прогрессирующей деформацией костей, несовершенным дентиногенезом, переломами, развивающимися в первый год жизни.
IV тип – наследуется аутосомно-доминантным путем. Характеризуется маленьким ростом, деформацией скелета, часто переломами костей, несовершенным дентиногенезом, нормальными склерами.
В течении несовершенного остеогенеза выделяют четыре стадии: латентную стадию, стадию патологических переломов, стадию глухоты и стадию остеопороза.
Как составная часть различных наследственных синдромов, несовершенный остеогенез может сочетаться с микроцефалией и катарактой; врожденными контрактурами суставов (синдром Брука) и др.
Симптомы несовершенного остеогенеза
Манифестация и тяжесть клинических проявлений несовершенного остеогенеза зависит от генетического типа заболевания.
При внутриутробной форме несовершенного остеогенеза в большинстве случаев дети появляются на свет мертворожденными. Более 80% живых новорожденных умирает на первом месяце жизни, из них более 60% – в первые дни. У детей с фетальной формой несовершенного остеогенеза отмечаются несовместимые с жизнью внутричерепные родовые травмы, синдром дыхательных расстройств, респираторные инфекции. Характерно наличие тонкой бледной кожи, истонченной подкожной клетчатки, общей гипотонии, переломов бедренной кости, костей голени, костей предплечья, плечевой кости, реже – переломов ключицы, грудины, тел позвонков, которые могут возникать внутриутробно или в процессе родовой деятельности. Все дети с внутриутробной формой несовершенного остеогенеза обычно умирают в течение первых 2-х лет жизни.
Поздняя форма несовершенного остеогенеза характеризуется типичной триадой симптомов: повышенной ломкостью костей, главным образом, нижних конечностей, синевой склер и прогрессирующей тугоухостью (глухотой). В раннем возрасте отмечается позднее закрытие родничков, отставание ребенка в физическом развитии, разболтанность суставов, атрофия мышц, подвывихи или вывихи. Переломы костей у ребенка с несовершенным остеогенезом могут возникать во время пеленания, купания, одевания ребенка, во время игр. Неправильное сращение патологических переломов часто приводит к деформации и укорочению костей конечностей. Переломы костей таза и позвоночника случаются реже. В старшем возрасте развиваются деформации грудной клетки и искривление позвоночника.
Несовершенный дентиногенез проявляется поздним прорезыванием зубов (после 1,5 лет), аномалиями прикуса; желтой окраской зубов («янтарные зубы»), их патологическим стиранием и легким разрушением, множественным кариесом. Вследствие выраженного отосклероза к 20-30 годам развиваются тугоухость и глухота. В постпубертатном периоде склонность к переломам костей уменьшается.
Сопутствующие проявления несовершенного остеогенеза могут включать пролапс митрального клапана, митральную недостаточность, чрезмерную потливость, камни в почках, пупочные и паховые грыжи, носовые кровотечения и др. Умственное и половое развитие детей при несовершенном остеогенезе не страдает.
Диагностика
Пренатальная диагностика позволяет выявить тяжелые формы несовершенного остеогенеза у плода с помощью акушерского УЗИ, начиная с 16 недели беременности. Иногда для подтверждения предположений проводится биопсия ворсин хориона и ДНК-диагностика.
В типичных случаях диагноз несовершенного остеогенеза ставится на основании клинико-анамнестических и рентгенологических данных. Обычно грубые морфологические и функциональные изменения выявляются на рентгенограммах трубчатых костей: выраженный остеопороз, истончение кортикального слоя, множественные патологические переломы с образованием костных мозолей и т. д.
Достоверность диагноза подтверждается гистоморфометрическим исследованием костной ткани, полученной во время пункции подвздошной кости, и структуры коллагена 1 типа в биоптате кожи. С целью выявления характерных для несовершенного остеогенеза мутаций выполняется молекулярно-генетический анализ.
В рамках дифференциальной диагностики несовершенного остеогенеза необходимо исключение рахита, хондродистрофии, синдрома Элерса-Данлоса.
Больные с несовершенным остеогенезом нуждаются в наблюдении детского травматолога-ортопеда, педиатра, стоматолога, детского отоларинголога, генетика.
Лечение несовершенного остеогенеза
Терапия несовершенного остеогенеза, главным образом, паллиативная, направленная на улучшение минерализации костной ткани; предотвращение переломов; физическую, психологическую и социальную реабилитацию.
Ребенку с несовершенным остеогенезом показан щадящий режим, курсы лечебной гимнастики, массажа, гидротерапии, физиотерапии (УФО, электрофорез солей кальция, индуктотермия, магнитотерапия). Из медикаментозных средств применяются поливитамины, витамин D, препараты кальция и фосфора. Для стимуляции синтеза коллагена назначается соматотропин, по завершении курса лечения которым показан прием стимуляторов минерализации костной ткани ( экстракта щитовидных желез скота, холекальциферола). Хорошие результаты при комплексной терапии несовершенного остеогенеза получены от применения бисфосфонатов, тормозящих разрушение костной ткани (памидроновой и золедроновой кислоты , ризедроната).
При переломах необходима тщательная репозиция костных отломков и гипсовая иммобилизация. При выраженных деформациях костей показано проведение хирургического лечения – корригирующей остеотомии с интрамедуллярным или накостным остеосинтезом.
Реабилитация детей, страдающих несовершенным остеогенезом, проводится группой специалистов: педиатром, детским ортопедом, физиотерапевтом, специалистом ЛФК, детским психологом и др. Детям может потребоваться ношение специальной ортопедической обуви, ортезов, стелек, корсетов.
Прогноз и профилактика
Дети с врожденной формой несовершенного остеогенеза погибают в первые месяцы и годы жизни от последствий множественных переломов и септических осложнений (пневмонии, отита, сепсиса). Поздняя форма несовершенного остеогенеза протекает более благоприятно, хотя и ограничивает качество жизни.
Профилактика сводится в основном к правильному уходу за ребенком, проведению лечебно-реабилитационных курсов, предупреждению бытовых травм. Наличие в семье больных с несовершенным остеогенезом служит прямым показанием к медико-генетическому консультированию.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 3 октября 2017; проверки требуют 18 правок.
Несоверше́нный остеогене́з (НО) (лат. osteogenesis imperfecta; иначе «несовершенное костеобразование», болезнь «хрустального человека», болезнь Лобштейна — Вролика) — группа генетических нарушений. Одно из заболеваний, характеризующееся повышенной ломкостью костей. Больные либо имеют недостаточное количество коллагена, либо его качество не соответствует норме. Так как коллаген — важный белок в структуре кости, это заболевание влечёт за собой слабые или ломкие кости.
Будучи генетическим нарушением, НО является аутосомно-доминантным дефектом, в большинстве переданным по наследству от родителей, однако, возможна и индивидуальная спонтанная мутация.
Типы[править | править код]
Существуют основные четыре типа НО. I тип наиболее частая и лёгкая форма, за которой следуют II, III и IV типы. Сравнительно недавно были классифицированы типы V, VI, VII и VIII которые разделяют те же клинические особенности что и 4-й, но каждый из них имеет уникальные гистологические и генетические данные.
| Тип | Описание | Ген | OMIM | Режим наследования |
| I | лёгкий | нулевой COL1A1 аллель | 166240 (IA), 166200 (IB) | аутосомно-доминантный, 60% de novo[2] |
| II | тяжёлый и зачастую смертельный в перинатальном периоде | COL1A1, COL1A2, | 166210 (IIA), 610854 (IIB) | аутосомно-рецессивный[3], ~100% de novo[2] |
| III | рассматривается, как прогрессивный и деформирующий | COL1A1, COL1A2 | 259420 | аутосомно-рецессивный[3], ~100% de novo[2] |
| IV | деформирующий, но с нормальной склерой | COL1A1, COL1A2 | 166220 | аутосомно-доминантный, 60% de novo[2] |
| V | клинические признаки соответствуют типу IV, но имеет и уникальные гистологические данные (“сетчато-подобный”) | неизвестен | 610967 | аутосомно-доминантный[2] |
| VI | клинические признаки соответствуют типу IV, но имеет и уникальные гистологические данные (“рыбья чешуя”) | неизвестен | 610968 (IVA) и (IVB) | неизвестен[2] |
| VII | связан с мутацией протеина хрящевых тканей | CRTAP | 610682 | аутосомно-рецессивный[2] |
| VIII | тяжелый и смертельный, связан с белком лейцин-пролин обогащенного протеогликана (Leprecan) | LEPRE1 | 610915 | аутосомно-рецессивный |
1-й тип[править | править код]
Коллаген нормального качества, но вырабатывается в недостаточных количествах.
- Кости легко ломаются, в особенности до пубертата
- Лёгкое искривление спины
- Слабость связочного аппарата суставов
- Пониженный мышечный тонус
- Обесцвечивание склер (глазного белка), обычно придающие им голубовато-карий цвет
- Ранняя потеря слуха у некоторых детей
- Слегка выступающие глаза
Также различают 1-й тип A и 1-й тип В по наличию или отсутствию несовершенного дентиногенеза (характеризуемый опаловыми зубами; отсутствует в IA, присутствует в IB). Помимо повышенного риска фатальных переломов костей, ожидаемая продолжительность жизни в пределах нормы.
2-й тип[править | править код]
Коллаген недостаточного количества или качества.
- Большинство случаев умирает на протяжении первого года жизни по причине дыхательной недостаточности или внутричерепного кровоизлияния,
- трудности с дыханием в связи с недоразвитыми лёгкими,
- тяжёлые деформации кости и невысокий рост.
2-й тип может быть далее разбит на подклассы A, B, C, различаемые радиографическим анализом длинной трубчатой кости и рёбер.
3-й тип[править | править код]
Коллаген в достаточных количествах, но недостаточного качества.
- Кости ломаются легко, иногда даже при рождении,
- деформации костей, часто тяжёлые,
- возможны проблемы с дыханием,
- невысокий рост, искривление позвоночника, иногда также бочковидная грудная клетка,
- слабость связочного аппарата суставов,
- слабый мускульный тонус в руках и ногах,
- обесцвечивание склер (глазных белков),
- иногда ранняя потеря слуха.
3-й тип выделяется из других классификаций будучи типом «Прогрессивной деформации», где новорожденный представляет лёгкие симптомы при рождении и развивает вышеуказанные симптомы в процессе жизни. Продолжительность жизни может быть нормальной, хотя и с тяжёлыми физическими препятствиями.
4-й тип[править | править код]
Коллаген достаточного количества, но недостаточно высокого качества.
- Кости ломаются легко, особенно до пубертата
- невысокий рост, искривления позвоночника и бочковидная грудная клетка,
- деформация костей в диапазоне от слабой до средней,
- ранняя потеря слуха.
Подобно 1-му типу, 4-й тип может быть далее разделён на подклассы IVA и IVB, которым характерно отсутствие (IVA) или наличие (IVB) несовершенного дентиногенеза.
Методы терапии[править | править код]
Так как НО является генетическим заболеванием, возможные формы терапии ограничиваются исключительно симптоматическими методами лечения.
В частности к ним принадлежат:
- остеосинтез штифтом,
- физиотерапия,
- терапия Бисфосфонатом.
витамин Д3
препараты кальция
Остеосинтез штифтом[править | править код]
При остеосинтезе штифтом изогнутая кость сначала многократно остеотомируется, чтобы затем бусообразно нанизывать костные сегменты на интрамедуллярный гвоздь. Сначала для этого использовались жёсткие штифты. В растущей кости, однако, такие штифты приходилось периодически заменять, так как кость однажды становилась длиннее штифта, вследствие чего штифт не был больше способен служить поддержкой для кости. Следовали фрактуры в этих незащищённых областях. Поэтому в 1963 году ортопедами был сконструирован выдвижной штифт. При росте кости два сегмента штифта выдвигаются друг из друга по принципу устройства подзорной трубы и как бы растут вместе с костью.
- Показан остеосинтез штифтом, людям с частыми переломами одной и той же кости, с ложными суставами, а также имеющим средние и тяжёлые смещение или функциональное нарушение суставов.
- Противопоказан в том числе при тяжёлом общем состоянии, сердечно-дыхательной недостаточности или отсутствии возможности закрепления штифта в кости из-за недостатка костной ткани.
Физиотерапия[править | править код]
Терапия Бифосфонатом[править | править код]
Примечания[править | править код]
- ↑ база данных Disease ontology (англ.) — 2016.
- ↑ 1 2 3 4 5 6 7 Steiner, RD; Pepin, M.G., Byers, P.H., Pagon, R.A., Bird, T.D., Dolan, C.R., Stephens, K., Adam, M.P. Osteogenesis Imperfecta. — 2005. — 28 января. — PMID 20301472.
- ↑ 1 2 Остеогенез несовершенный // 1. Малая медицинская энциклопедия. — Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — Советская энциклопедия. — 1982—1984 гг (рус.). — М.
Ссылки[править | править код]
- Несовершенный остеогенез: постсоюзная территория
- Osteogenesis imperfecta на MedLinks.ru
- «Хрустальные дети» с «хрупким» заболеванием
- «Хрупкие дети» – официальный сайт Первой общественной организации помощи детям с НО
Источник
Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
Данная гетерогенная группа генетических заболевний (разделенная на четыре типа, определенных Силленсом (Sillence): I, II, или врожденный, III и IV) характеризуется выраженной хрупкостью костей, ведущей к патологической оссификации и множественным переломам. Имеются следующие признаки при каждом их четырех типов заболевания.
• Тип I не характеризуется пренатальными изменениями, и диагноз устанавливается после рождения, когда начинают появляться деформации конечностей.
• Тип II является наиболее тяжелой формой. Она проявляется множественными нарушениями развития скелета, такими как укорочение и угловые искривления костей и появление выступов на их поверхностях вследствие множественных переломов, а также деминерализацией костей свода черепа, узкой и колоколообразной грудной клеткой в результате переломов ребер и уменьшением двигательной активности плода.
• Тип III является менее тяжелым, чем тип II, и обычно проявляется множественными переломами при рождении с развитием прогрессирующих деформаций костей от неонатального периода до подросткового возраста. Эта форма заболевания диагностируется начиная со второго триместра беременности при типе NIC.
• Тип IV характеризуется наименее выраженными проявлениями нарушений и пренатально не определяется. Заболевание обычно сопровождается преждевременным развитием остеопороза в возрасте 40-50 лет.
Синонимы. Врожденный несовершенный остеогенез, синдром Ван дер Хове (Van der Hoeve), болезнь Лобстейна (Lobstein), меловые хрупкие кости, болезнь хрупких костей, болезнь Вролика (Vrolik).
Распространенность. Частота возникновения составляет 0,4 на 10 000 новорожденных и примерно половина из них (0,19 на 10 000) имеет тип II.
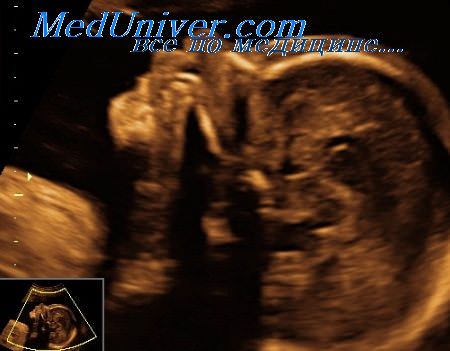
Этиология. В большинстве случаев возникновение типов I и IV обусловлено аутосомно-доминантным наследованием. Тип II формируется в результате новой доминантной мутации (с несколькими сообщениями об аутосомно-рецессивном наследовании), и тип III образуется аутосомно-рецессивно или аутосомно-доминантно.
Риск рецидива. Зависит от формы наследования заболевания. В целом риск рецидива составляет от 2 до 5% для типа II в тех случаях, когда это не вызвано спонтанной мутацией.
Диагностика. Эхографические признаки, которые могут выявляться, особенно при типе II, включают увеличение ширины, укорочение и переломы длинных трубчатых костей, которые имеют выступы на поверхности, образованные костными мозолями; снижение оссификации костей свода черепа, улучшение визуализации внутричерепных структур, уменьшенную в размерах и колоколообразную грудную клетку, аномальную форму мозгового черепа; широкие ребра неравномерной толщины, искривление длинных трубчатых костей и патологию лицевых структур. Типичным признаком несовершенного остеогенеза II типа является возможность визуализировать оба кортикальных слоя костей. Обычно дистальный по отношению к датчику слой попадает в зону «акустической тени», формирующейся позади проксимального слоя.
Однако при несовершенном остеогенезе этот феномен отсутствует вследствие выраженной деминерализации (аналогичные признаки могут выявляться при ахондрогенезе и гипофосфатазии). Большинство аномалий может быть выявлено пренатально с помощью эхографии начиная с первой половины второго триместра беременности. Нормальные эхографические данные у пациенток из группы риска по развитию заболевания у плода полностью не исключают наличие диагноза. В большинстве случаев если в пренатальном периоде переломы не возникают, то диагноз обычно устанавливается только после рождения.
Генетические нарушения. Причиной заболевания являются мутации одного из двух генов, ответственных за синтез коллагена I типа (COL1А1 и COL1A2).
Патогенез. Дефекты синтеза, организации и химического строения коллагена I типа, который присутствуете коже, связках, сухожилиях деминерализованных костях и дентине, являются причиной снижения минерализации и возникновения хрупкости костей.
Сочетанные аномалии. Кифосколиоз, глухота, мышечная гипотония, паховые грыжи, гидроцефалия, водянка и задержка внутриутробного развития.
Дифференциальный диагноз. Гипофосфатазия (инфантильная форма), ахондрогенез и другие заболевания, сопровождающиеся нанизмом и укорочением конечностей.
Прогноз. Несовершенный остеогенез II типа всегда летален, и наиболее частыми причинами гибели являются, вероятно, внутричерепные кровоизлияния и дыхательная недостаточность. Другие формы развиваются после рождения и сопровождаются прогрессирующими деформациями длинных трубчатых костей и наличием низкого роста.
Акушерская тактика. Вследствие летальности заболевания при диагностике несовершенного остеогенеза II типа прерывание беременности может быть предложено на любом сроке гестации. Для других форм прерывание предлагается до наступления периода жизнеспособности плода. Если отдается предпочтение пролонгированию беременности, стандартная пренатальная тактика ведения не изменяется.
– Также рекомендуем “Синдром Пена-Шокейра. Пентада Кантрелла”
Оглавление темы “Врожденные синдромы плода”:
1. Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
2. Синдром летального птеригума. Лиссэнцефалия I типа
3. Синдром Мекеля. Диагностика и прогноз при синдроме Мекеля
4. Мезомелическая дисплазия. Диагностика и прогноз мезомелической дисплазии
5. Микроцефалический первичный нанизм. Синдром Неу-Лаксова
6. Синдром Нунан у плода. Секвенция маловодия
7. Несовершенный остеогенез. Диагностика и прогноз несовершенного остеогенеза у плода
8. Синдром Пена-Шокейра. Пентада Кантрелла
9. Синдром Пфайффера. Диагностика и прогноз синдрома Пфайффера
10. Синдром Поланда. Секвенция Поттер и синдром prune-belly
Источник